FDA Inspection of Generic Manufacturing Facilities: What to Expect in 2025

When the FDA shows up at your generic drug manufacturing facility, it’s not a surprise visit-it’s a routine check. But that doesn’t mean you can wing it. Every year, hundreds of facilities across the U.S. and overseas get inspected to make sure they’re following Current Good Manufacturing Practices (CGMP). These rules, laid out in 21 CFR Part 211, aren’t suggestions. They’re the law. And if you’re making pills, injections, or ointments for the American market, the FDA has the right to walk through your plant anytime-announced or not.
Why the FDA Inspects Generic Drug Factories
The FDA doesn’t inspect generic drug facilities just to check boxes. They’re protecting patients. A single batch of contaminated or improperly labeled medicine can cause real harm. That’s why the agency uses a risk-based system to pick which sites get visited. Facilities that make high-risk products, have had past violations, or got a tip about problems are more likely to be targeted. But even the cleanest plants aren’t immune. Routine inspections happen across the board.
More than 90% of inspections find facilities in acceptable compliance. That’s not luck-it’s preparation. Companies that treat quality like a daily habit, not a checklist, rarely get caught off guard. The FDA’s goal isn’t to shut you down. It’s to make sure your medicine works the way it’s supposed to, every time.
Types of Inspections You Might Face
Not all FDA inspections are the same. There are four main types, and each has a different focus.
- Pre-Approval Inspections (PAIs) happen before a new generic drug gets approved. The FDA wants to confirm your facility can actually make the product you described in your application. They’ll check if your equipment, processes, and lab methods match what you submitted. If your stability samples aren’t stored where you said they’d be, or your analytical methods don’t match the filed ones, your approval could stall.
- Routine Surveillance Inspections are the most common. These happen every 2-3 years for most facilities. The FDA uses a 6-system approach: Quality, Facilities & Equipment, Materials, Production, Packaging & Labeling, and Laboratory Control. The Quality System is always reviewed-no exceptions. They’ll pick two or three more systems to dig into based on risk.
- For-Cause Inspections are triggered by specific red flags: a spike in consumer complaints, a whistleblower tip, or a product recall. These are intense. Investigators zero in on the problem area but still look at the bigger picture. One bad process doesn’t mean the whole system is broken-but it will be scrutinized.
- Follow-Up Inspections happen after a warning letter or an unacceptable finding. The FDA comes back to see if you fixed what they flagged. If you didn’t, they might take legal action.
What the FDA Investigators Look For
Inspectors don’t just walk around. They dig into your records, watch your people work, and ask questions that feel like a pop quiz. Here’s what they’re really checking:
- Quality Control Unit (21 CFR 211.22): Do you have a team that’s independent from production and empowered to stop shipments? If quality is just another department, you’re already in trouble.
- Process Validation: Can you prove your manufacturing process consistently produces the right product? Not just once. Every batch. You need data-thousands of data points-to show it’s repeatable.
- Equipment Qualification: Is your mixer calibrated? Is your autoclave validated? Are your sensors working? If you can’t show proof, you’re not compliant.
- Supplier Qualification: Where do your raw materials come from? Do you test them? Do you audit your suppliers? If you’re buying active ingredients from a vendor you’ve never visited, that’s a red flag.
- Testing Protocols: Are your lab methods accurate? Are your analysts trained? Are your reference standards current? The FDA will pull random samples and compare your results to theirs.
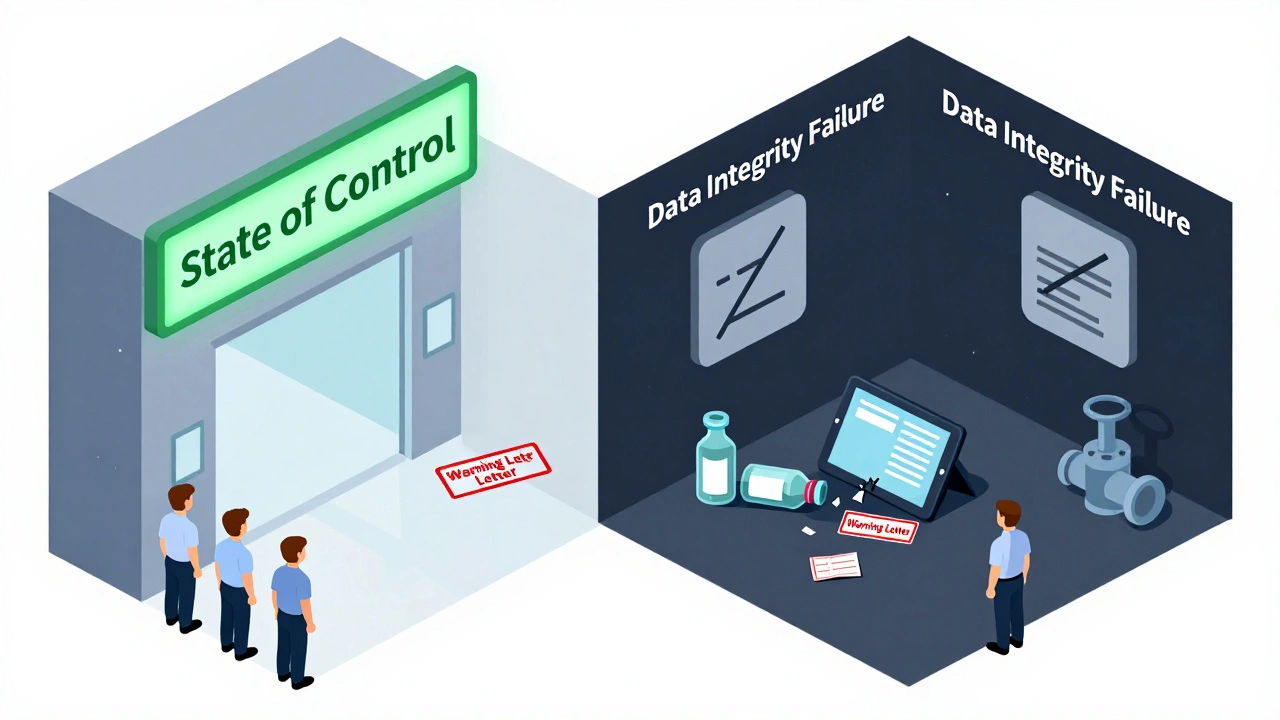
- Data Integrity: This is huge now. Are your records real? Are timestamps locked? Are deletion logs turned on? If someone altered a result, deleted a batch record, or used a spreadsheet without controls, you’re in serious trouble.
- Stability Testing: Where are your samples stored? Are they kept at the exact temperature and humidity you filed with the FDA? If your stability data doesn’t match your claims, your drug’s shelf life is invalid.
The FDA doesn’t need to find a major violation to shut you down. A pattern of small issues-missing signatures, incomplete training logs, unapproved changes-adds up. They call it “a state of control.” If you’re not in control, you’re not compliant.

What Happens During the Inspection
You’ll get a notice-sometimes weeks in advance, sometimes the day before. When the team arrives, they’ll introduce themselves and ask for your inspection area. That’s not a suggestion. You need a clean, quiet room with a table, chairs, power outlets, and internet. No lunchroom. No break area. They’ll want to see your SOPs, batch records, validation reports, and training files.
They’ll tour your facility. They’ll watch operators load a machine. They’ll ask why a valve is labeled a certain way. They’ll open your warehouse and pick a random container. They’ll check if your environmental monitoring logs match your actual conditions.
At the end of the day, they’ll give you Form FDA 483. This isn’t a final verdict. It’s a list of observations-things they saw that don’t meet CGMP standards. Each item references a specific regulation, like 21 CFR 211.110 for missing batch record reviews. You have 15 business days to respond. Your response isn’t just an apology. It’s a plan: what you found, why it happened, how you fixed it, and how you’ll prevent it again.
The 483 Response: Your Best Shot at Avoiding a Warning Letter
Most companies panic when they get a 483. Bad move. The FDA expects you to respond. What they don’t want is vague answers like “we’ve retrained staff.” That’s not a fix. That’s noise.
A strong response includes:
- Specific root cause analysis
- Corrective actions with timelines
- Preventive actions that change systems, not just people
- Evidence-photos, updated SOPs, training records, validation reports
If your response is weak, the FDA may issue a warning letter. That’s public. It’s on their website. It can trigger import alerts, delay approvals, and scare off customers. But even then, you’re not done. The FDA now offers Post-Warning Letter Meetings (PWLMs), a formal chance to discuss your corrective actions before they escalate. Use it.
How to Prepare-Before They Even Show Up
You don’t prep for an inspection. You prep for a life of quality.
- Simulate inspections: Do mock audits every quarter. Bring in someone from another site or hire a consultant. Act like the FDA is walking in tomorrow.
- Keep your facility clean: A dusty floor, a broken light, a cluttered lab bench-they all signal neglect. The FDA notices everything.
- Train your staff: Everyone, from the janitor to the CEO, should know what an FDA inspector is and what to say (and not say). “I don’t know” is fine. “That’s not my job” is not.
- Document everything: If it wasn’t written down, it didn’t happen. Every change, every deviation, every test-documented, reviewed, signed.
- Use the PreCheck program: Launched in 2024, this is your chance to get feedback before you build or scale. Submit a Type V Drug Master File (DMF) with your facility layout, quality system design, and validation plans. The FDA will give you early input. It’s free. It’s smart.
Companies with mature quality cultures don’t get surprised. They don’t get 483s. They get constructive feedback. The FDA respects them. That’s the goal.
What Comes After the Inspection
The FDA writes an Establishment Inspection Report (EIR). This is their official record. It says whether you’re in “a state of control” or not. It’s not public, but it’s shared internally. If you’re flagged, it affects future inspections.
If you passed, great. But don’t relax. The FDA may come back sooner than you think. If you had issues, they’ll monitor you closely. Your next inspection might be in 6 months, not 3 years.
The FDA’s goal isn’t to punish. It’s to improve. They’ve spent years refining their approach. They’re not just looking for compliance-they’re looking for quality culture. If your team owns quality every day, you won’t just pass inspections. You’ll build trust.
Final Thought: It’s Not About Avoiding the FDA
It’s about working with them. The FDA doesn’t want to be your enemy. They want you to make safe, effective medicine. The system works when you’re transparent, thorough, and honest. Don’t treat inspections like a threat. Treat them like a chance to prove you’re doing it right.
Most facilities pass. The ones that don’t? They didn’t fail because of one mistake. They failed because they stopped caring.
Can the FDA inspect my facility without notice?
Yes. While many inspections are scheduled, the FDA has the legal right to conduct unannounced inspections at any time, especially for facilities with higher risk profiles or past compliance issues. Unannounced visits are becoming more common as part of the agency’s risk-based strategy.
What happens if I don’t respond to an FDA 483?
Failing to respond within 15 business days is seen as non-cooperation. The FDA may proceed to issue a warning letter without waiting for your input. A warning letter is public, can block product approvals, and may trigger import alerts or regulatory actions.
How long does an FDA inspection usually last?
Most routine inspections last 3-7 days, depending on facility size and complexity. Pre-Approval Inspections (PAIs) can take longer-up to 10 days or more-because they involve deep review of application data and process validation. For-cause inspections may be shorter but more intense.
What’s the difference between a 483 and a warning letter?
An FDA 483 is a list of observations made during the inspection-it’s not a formal enforcement action. A warning letter is a legal notice that the FDA has determined your facility is in violation of federal law. It follows if your 483 response is inadequate or if violations are severe.
Can I appeal an FDA inspection finding?
You can’t appeal the 483 itself, but you can respond to it with detailed corrective actions. If a warning letter is issued, you can request a formal meeting with the FDA to discuss your response. In rare cases, you may request a hearing through the agency’s administrative process, but this is uncommon and legally complex.
Does the FDA inspect foreign generic drug manufacturers too?
Yes. The FDA inspects foreign facilities at the same standards as U.S. sites. In fact, over half of the generic drugs sold in the U.S. are made overseas. The agency has offices in India, China, and other key manufacturing regions and conducts thousands of international inspections annually.
How does the PreCheck program help manufacturers?
The PreCheck program, launched in 2024, lets manufacturers submit detailed facility and process plans early-during design or construction. The FDA reviews them and gives feedback before production starts. This reduces the risk of costly redesigns or delays during Pre-Approval Inspections. It’s a proactive way to avoid surprises.
What’s the most common reason facilities fail FDA inspections?
The most common failure point is poor data integrity. This includes missing or altered records, lack of audit trails, unapproved changes to methods, and failure to validate software systems. The FDA now trains inspectors specifically to spot these issues, and they’re prioritized over minor paperwork errors.
Do I need a quality unit if I’m a small generic manufacturer?
Yes. The FDA requires a qualified quality unit regardless of company size. Even small manufacturers must have a team or designated person with authority to review and approve all quality-related decisions, including batch release, deviations, and investigations. You can’t outsource this responsibility.
What’s the best way to train staff for an FDA inspection?
Train them to be honest, calm, and clear. Don’t rehearse scripted answers. Instead, teach them to say, “I’ll get the right person,” if they don’t know. Role-play real scenarios: what happens if an inspector asks about a batch record? What if they find a broken seal? Practice responses that show ownership, not defensiveness.




Comments (15)
Audrey Crothers
11 Dec 2025
Just had an FDA inspection last month-passed with zero 483s! The key? We stopped treating compliance like a yearly chore and started living it. Every morning, we do a 5-minute quality huddle. Simple, but it works.
Also, the PreCheck program? Total game-changer. Got feedback before we even built the new line. Saved us months.
Stacy Foster
13 Dec 2025
They’re lying. The FDA doesn’t care about patients-they care about control. Every time they shut down a foreign plant, another Big Pharma monopoly gets stronger. You think they inspect because of safety? Nah. They do it because they can.
And don’t get me started on the ‘PreCheck’ scam-just another way to make you pay for their bureaucracy. Wake up.
They’re not here to help. They’re here to intimidate.
Robert Webb
15 Dec 2025
There’s a lot of truth here, especially about data integrity being the #1 failure point. I’ve seen too many small labs fail because they used Excel spreadsheets without audit trails-no version control, no password protection, no logs. One person changes a number, no one knows who or when.
It’s not about being paranoid-it’s about being traceable. The FDA doesn’t need to find a huge violation to shut you down. One missing signature on a calibration log, repeated across 12 batches? That’s a pattern. And patterns mean you’re not in control.
Also, training staff to say ‘I’ll get the right person’ is gold. Don’t guess. Don’t bluff. Don’t pretend. That’s what separates the survivors from the cautionary tales.
Reshma Sinha
15 Dec 2025
As someone who’s worked in both US and Indian GMP facilities, I can tell you-the standards are identical. But the culture? Totally different.
In the US, they treat compliance like a religion. In India, it’s often seen as a cost center. That’s why so many foreign sites get flagged-not because they’re bad, but because they’re reactive, not proactive.
But here’s the good news: the ones who invest in real quality culture? They fly under the radar and get respect. It’s not about fear. It’s about pride in what you make.
nikki yamashita
17 Dec 2025
YESSS. PreCheck is legit. My boss was skeptical, but we applied last year and got feedback on our lab layout before we spent $2M on equipment. Now we’re all like, why didn’t we do this sooner?
Also, the ‘I don’t know’ rule? Lifesaver. My team used to panic and make stuff up. Now they just say ‘let me find out’ and come back with the answer. FDA loves that.
wendy b
18 Dec 2025
Actually, the FDA’s 6-system approach is outdated. They’re still using 1970s logic in a 2025 digital world. If you’re not using AI-driven anomaly detection in your QC lab, you’re already behind. And don’t even get me started on how they still demand paper-based batch records in 2025.
Also, ‘quality unit’? Please. In a 10-person shop, the CEO should be the quality unit. Stop pretending you need a whole department. It’s performative bureaucracy.
Laura Weemering
20 Dec 2025
...and yet...
...and yet...
...and yet...
...the FDA... doesn’t... understand... the... human... cost...
...of... this... relentless... scrutiny...
...when... the... same... person... is... responsible... for... validation... records... and... batch... release... and... training... logs... and... they’ve... been... working... since... 5am...
...and... the... inspector... asks... ‘why... is... this... signature... missing?’...
...and... they... don’t... see... the... 14-hour... day... the... 3... missed... birthdays... the... burnout...
...they... just... see... a... box... unchecked...
...and... we... are... not... machines...
...we... are... people... trying... to... make... medicine...
...but... the... system... treats... us... like... glitches...
...and... I... am... so... tired...
...and... I... don’t... know... how... much... longer... I... can... keep... pretending... this... is... about... safety...
...and... not... control...
...and... not... fear...
...and... not... punishment...
...and... not... the... quiet... death... of... a... thousand... tiny... boxes...
Rob Purvis
22 Dec 2025
One thing no one talks about: the emotional toll on QA teams. I’ve seen people cry after a 483. Not because they failed, but because they knew they’d spent 18 months building something perfect, and one broken seal, one missed signature, one untrained temp worker-it all unraveled.
It’s not just about systems. It’s about people who care. And the system doesn’t reward care. It rewards documentation.
So we document. We over-document. We create redundant checks for checks. Because if you don’t, the FDA sees negligence. And they’re right to. But it’s exhausting.
And honestly? The FDA inspectors I’ve met? Most of them are just trying to do their job. They’re not villains. They’re tired too. We just need to stop treating them like enemies and start treating them like partners. Even if it’s hard.
Lawrence Armstrong
22 Dec 2025
PreCheck is genius. We used it for our new lyophilization line. FDA gave us 3 tweaks before we even installed the dryer. Saved us $400K and 6 months.
Also, if you’re still using paper logs, you’re not just outdated-you’re dangerous. Digital systems with audit trails aren’t optional anymore. They’re basic hygiene.
And yes, data integrity is the #1 killer. Seen it too many times. Someone ‘corrects’ a value because ‘it looked wrong.’ Nope. That’s fraud. Even if you meant well.
Don’t be that person.
Donna Anderson
23 Dec 2025
my boss said we dont need to worry about the 483 until they show up but now i get it its not about waiting its about living it every day like breathing
we started doing 5 min quality checks before shift start and its changed everything
no more panic just pride
sandeep sanigarapu
24 Dec 2025
Inspection in India: same rules, different reality. We have 12 people in QA, 300 in production. The FDA expects the same standards as a 500-person US plant. It’s not fair. But it’s the law.
So we train everyone like they’re QA. Even the warehouse guy knows what a deviation log is. We make it part of the culture, not a department. It works.
And yes, foreign plants get inspected more now. But we’re ready. Because we have to be.
Ashley Skipp
24 Dec 2025
Why do we even bother with all this paperwork when 90% of generics are made overseas and the FDA can’t even inspect them all
it’s a joke
they pick on small shops because they’re easy targets
big pharma gets a pass
you think they care about your batch record
they care about their stock price
Levi Cooper
26 Dec 2025
Why are we letting foreign countries make our medicine? The FDA inspects them, sure-but they’re not American. They don’t care about our patients like we do. We used to make 80% of our generics at home. Now it’s 10%.
This isn’t about compliance. This is about national security.
And don’t tell me ‘cost efficiency.’ What’s the cost of a contaminated batch? Lives. And we’re outsourcing our safety to a country with different values.
Wake up, America.
Nathan Fatal
26 Dec 2025
What’s really being asked here isn’t whether you’re compliant-it’s whether you’re worthy. The FDA doesn’t want perfect paperwork. They want a culture that says, ‘We don’t cut corners because we know someone’s life depends on it.’
That’s why the best facilities don’t fear inspections. They welcome them. Because they know their people, their systems, their values are solid.
It’s not about passing a test. It’s about living a promise.
And if you’ve forgotten that promise? No amount of SOPs will save you.
Start with why.
Robert Webb
28 Dec 2025
Reshma and Lawrence hit it right. The real difference isn’t in the checklist-it’s in the silence between the lines. The quiet moment when someone stops to double-check a label because ‘it just felt off.’ That’s not in any regulation. That’s culture.
And that’s what the FDA senses, even if they can’t measure it.
That’s why some sites get a nod and a smile. Others get a 483. Not because of what’s written-but because of what’s felt.